publications
publications by categories in reversed chronological order. generated by jekyll-scholar.
Alternative sources for our publications:
2025
2025
- Splice-switching antisense oligonucleotides correct cryptic exon inclusion and restore SDCCAG8 protein in Bardet-Biedl SyndromeKelleen E McEntee, Bailey L McCurdy, Austin Larson, and 8 more authors2025
Bardet-Biedl Syndrome (BBS) is a ciliopathy often associated with progressive blindness and obesity. A patient presenting with BBS was discovered to have two mutations within 55bp of each other in intron 7 of SDCCAG8 (BBS16). One of the biallelic mutations, c.740+356C>T, causes inclusion of cryptic exon(s) containing premature termination codons, while c.740+301G>A has not been characterized. We hypothesized that antisense oligonucleotides (ASOs) complementary to the patient’s mutations or to the cryptic exon splice sites would correct the splicing of SDCCAG8 between exons 7 and 8 to prevent cryptic exon inclusion and restore SDCCAG8 expression. We systematically screened 20nt-long ASOs tiled across each mutation and ASOs targeting the 3′ splice sites of the cryptic exons in patient-derived fibroblasts, using RT-PCR assays to assess exon 7 and 8 splicing. We identified one ASO for each mutation and a cryptic exon-targeting ASO that restored the splicing pattern to that observed in an unaffected cell line. Lead ASOs were further investigated through RT-PCR, RNA sequencing, and western blotting to confirm ASO-mediated restoration of wild-type transcript and protein. Notably, ASO 20, which targets the cryptic exon 7a/7a′ splice site rather than patient-specific mutations, achieved the greatest rescue effect, increasing exon 7-8 splicing from 0% to an average of 26% and restoring SDCCAG8 protein from undetectable levels to approximately 40% of wild-type expression. This mutation-agnostic approach could benefit multiple patients with cryptic exon inclusion in this region of SDCCAG8, expanding therapeutic impact beyond traditional N-of-1 ASO strategies. These findings establish a molecular foundation for clinical development of ASO therapy for BBS caused by SDCCAG8 splicing defects.
- Serine recombinases are conserved genetic markers of antiphage defense systemsShelby E Andersen, Joshua M Kirsch, Navtej Singh, and 4 more authors2025
Abstract Antiphage defense systems confer bacteriophage (phage) resistance in bacteria. Renewed interest in phage therapy indicates a need to understand the breadth and molecular mechanisms of antiphage defenses. Traditionally, strategies to identify antiphage defenses lack throughput or are biased toward model bacterial. Herein, we developed a bioinformatic pipeline that uses a serine recombinase to identify known and unknown antiphage defense systems. Using this approach to query reference genomes and metagenomes, we show that serine recombinase genes are genetically linked to antiphage defense systems and serve as bait for finding these systems across diverse bacterial phyla. Using co-transcription predictions and statistical analysis of protein domain abundances, we experimentally validated our informatic approach by discovering that KAP P-loop NTPases are fused to putative antiphage effector domains. Our work shows that serine recombinases are a reliable genetic marker for the discovery of antiphage defenses across bacterial phyla.
-
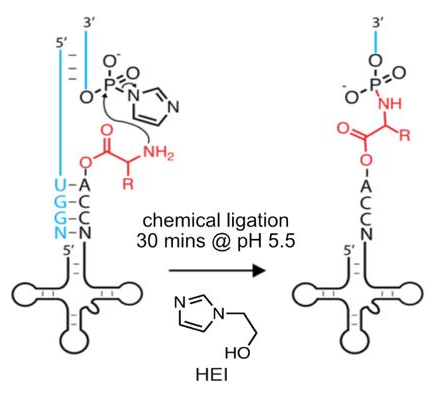 Nanopore sequencing of intact aminoacylated tRNAsLaura K White, Aleksandar Radakovic, Marcin P Sajek, and 5 more authors2025
Nanopore sequencing of intact aminoacylated tRNAsLaura K White, Aleksandar Radakovic, Marcin P Sajek, and 5 more authors2025Abstract The intricate landscape of tRNA modification presents persistent analytical challenges, which have impeded efforts to simultaneously resolve sequence, modification, and aminoacylation state at the level of individual tRNAs. To address these challenges, we introduce “aa-tRNA-seq”, an integrated method that uses chemical ligation to sandwich the amino acid of a charged tRNA in between the body of the tRNA and an adaptor oligonucleotide, followed by high throughput nanopore sequencing. Our approach reveals the identity of the amino acids attached to all tRNAs in a cellular sample, at the single molecule level. We describe machine learning models that enable the accurate identification of amino acid identities based on the unique signal distortions generated by the interactions between the amino acid in the RNA backbone and the nanopore motor protein and reader head. We apply aa-tRNA-seq to characterize the impact of the loss of specific tRNA modification enzymes, confirming the hypomodification-associated instability of specific tRNAs, and identifying additional candidate targets of modification. Our studies lay the groundwork for understanding the efficiency and fidelity of tRNA aminoacylation as a function of tRNA sequence, modification, and environmental conditions.
- Big1 is a cell cycle regulator linking cell size to basal body numberAlexander J Stemm-Wolf, Adam W J Soh, Lisa E Mitchell, and 9 more authors2025
Cell size control in dividing cells coordinates cell growth with cell division. In the ciliated protozoan, Tetrahymena, there is a tight link between cell size and the cytoskeletal assemblies at the cell cortex organized around basal bodies (BBs). BBs dictate the distribution of ciliary units governing cell motility and are organized into 18-22 ciliary rows. The number of BBs per cell remains remarkably consistent even when the number and lengths of ciliary rows vary. big1-1 mutant cells are large and have elevated numbers of BBs, providing a system to investigate links between BB number and cell size control. We discovered BIG1 encodes a protein with an RRM3 RNA-binding domain similar to the fission yeast meiotic entry gene, mei2 . The big1-1 mutation is a predicted null allele. By extending the duration of specific cell cycle stages conducive to new BB assembly, big1-1 promotes cell size increases through BB amplification. In contrast, excess Big1 protein localizes to BBs and drives cells into premature cell division, resulting in small cells with fewer BBs. Thus, Tetrahymena Big1 localizes to BBs and controls cell cycle progression, indicating BBs and Big1 link cell growth to the cell division cycle.
-
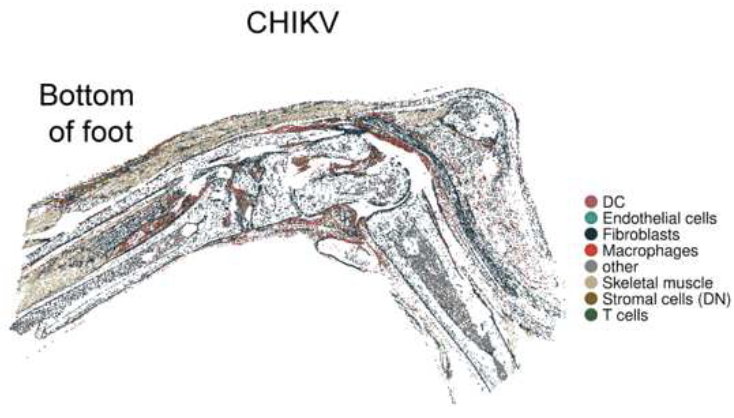 Chikungunya virus persists in joint associated macrophages and promotes chronic diseaseThomas Morrison, Kristen Zarrella, Ryan Sheridan, and 10 more authors2025
Chikungunya virus persists in joint associated macrophages and promotes chronic diseaseThomas Morrison, Kristen Zarrella, Ryan Sheridan, and 10 more authors2025Abstract Arthritogenic alphaviruses including chikungunya, Mayaro, and Ross River viruses cause long-lasting musculoskeletal pain and inflammation. However, mechanisms driving chronic disease remain poorly understood. Here, we investigated joint-associated tissues in alphavirus-infected mice at a late stage of infection. Utilizing scRNA-seq, spatial transcriptomics, and flow cytometry we identified an accumulation of inflammatory macrophages in joint-associated tissues with elevated Tnf, Nlrp3, Il1b, and H2-Aa expression, and these cells harbored CHIKV RNA. Moreover, we identified an accumulation of CD4+ T cells in joint-associated tissues, which express Ifng. Depletion of CD4+ T cells diminished MHC-II expression on joint macrophages, highlighting their potential role in inflammation. In addition, treatment with a small molecule inhibitor of CHIKV replication during chronic disease reduced viral RNA and joint inflammation, suggesting that viral RNA replication promotes chronic joint disease. Our data suggest that macrophages harbor replicating viral RNA and contribute to the sustained joint inflammation associated with chronic alphavirus disease.
-
 Systematic identification and characterization of eukaryotic and viral 2A peptide-bond-skipping sequencesDeviyani M Rao, Emma R Horton, Chloe L Barrington, and 10 more authors2025
Systematic identification and characterization of eukaryotic and viral 2A peptide-bond-skipping sequencesDeviyani M Rao, Emma R Horton, Chloe L Barrington, and 10 more authors20252A peptides are 18- to 22-amino-acid sequences that cause an unusual co-translational peptide-bond-skipping event. Initially discovered in viruses, they allow multiple proteins to be produced from a single open reading frame. Despite their utility, their evolutionary prevalence and sequence diversit …
-
 Systematic analysis of nonsense variants uncovers peptide release rate as a novel modifier of nonsense-mediated mRNA decayDivya Kolakada, Rui Fu, Nikita Biziaev, and 10 more authorsCell Genom., 2025
Systematic analysis of nonsense variants uncovers peptide release rate as a novel modifier of nonsense-mediated mRNA decayDivya Kolakada, Rui Fu, Nikita Biziaev, and 10 more authorsCell Genom., 2025The phenotypic impact of nonsense variants is determined by nonsense-mediated mRNA decay (NMD), which degrades transcripts with premature termination codons (PTCs). Despite the clinical importance of nonsense variants, transcript-specific and context-dependent variations in NMD activity remain poorly understood. Here, we show that the amino acid preceding the PTC strongly influences NMD activity. Glycine codons promote robust NMD efficiency and show striking enrichment before PTCs but are depleted before normal termination codons. Glycine-PTC enrichment is particularly pronounced in genes tolerant to loss-of-function variants, suggesting efficient elimination of truncated proteins from nonessential genes. We further demonstrate that the peptide release rate during translation termination is an important determinant of NMD activity. We propose a "window of opportunity" model where translation termination kinetics modulate NMD activity. By revealing how sequence context shapes NMD activity through translation termination dynamics, our findings provide a mechanistic framework for improved clinical interpretation of nonsense variants.
- Activated polyreactive B cells are clonally expanded in autoantibody positive and patients with recent-onset type 1 diabetesCatherine A Nicholas, Fatima A Tensun, Spencer A Evans, and 7 more authorsCell Rep., 2025
Autoreactive B cells play an important but ill-defined role in autoimmune type 1 diabetes (T1D). We isolated pancreatic islet antigen-reactive B cells from the peripheral blood of non-diabetic autoantibody-negative first-degree relatives, autoantibody-positive, and recent-onset T1D donors. Single-cell RNA sequencing analysis revealed that islet antigen-reactive B cells from autoantibody-positive and T1D donors had altered gene expression in pathways associated with B cell signaling and inflammation. Additionally, BCR sequencing uncovered a similar shift in islet antigen-reactive B cell repertoires among autoantibody-positive and T1D donors where greater clonal expansion was also observed. Notably, a substantial fraction of islet antigen-reactive B cells in autoantibody-positive and T1D donors appeared to be polyreactive, which was corroborated by analysis of recombinant monoclonal antibodies. These results expand our understanding of autoreactive B cell phenotypes during T1D and identify unique BCR repertoire changes that may serve as biomarkers for increased disease risk.
- B cells shape naïve CD8 T cell programmingCameron Manes, Miguel Guerrero Moreno, Jennifer Cimons, and 14 more authorsJ. Clin. Invest., 2025
The presence of B cells is essential for the formation of CD8 T cell memory after infection and vaccination. In this study, we investigated whether B cells influence the programming of naïve CD8 T cells prior to their involvement in an immune response. RNA sequencing indicated that B cells are necessary for sustaining the FOXO1-controlled transcriptional program, which is critical for their homeostasis. Without an appropriate B cell repertoire, mouse naïve CD8 T cells exhibit a terminal, effector-skewed phenotype, which significantly impacts their response to vaccination. A similar effector-skewed phenotype with reduced FOXO1 expression was observed in naïve CD8 T cells from human patients undergoing B cell-depleting therapies. Furthermore, we show that patients without B cells have a defect in generating long-lived CD8 T cell memory following COVID vaccination. In summary, we demonstrate that B cells promote the quiescence of naïve CD8 T cells, poising them to become memory cells upon vaccination.
- GPR182 is a lipoprotein receptor for dietary fat absorptionZhiwei Sun, Robert J Torphy, Emily N Miller, and 16 more authors2025
The lymphatic system plays a central role in lipid absorption, which transports chylomicrons from the small intestine to the circulation. However, the molecular mechanism by which chylomicrons get into the intestinal lymphatics is unknown. Here we demonstrated that GPR182, a receptor in lymphatic endothelial cells (LECs), mediates dietary fat absorption. GPR182 knockout mice are resistant to dietary-induced obesity. GPR182 ablation in mice leads to poor lipid absorption and thereby a delay in growth during development. GPR182 binds and endocytoses lipoproteins broadly. Mechanistically, loss of GPR182 prevents chylomicrons from entering the lacteal lumen of the small intestine. GPR182 blockage with a monoclonal antibody (mAb) protects mice from dietary induced obesity. Together, our study identifies GPR182 as a lipoprotein receptor that mediates dietary fat absorption.
2024
2024
- Co-regulator activity of Mediator of DNA Damage Checkpoint 1 (MDC1) is associated with DNA repair dysfunction and PARP inhibitor sensitivity in lobular carcinoma of the breastJoseph L Sottnik, Madeleine T Shackleford, Camryn S Nesiba, and 10 more authors2024
Invasive lobular carcinoma of the breast (ILC) are typically estrogen receptor α (ER)-positive and present with biomarkers of anti-estrogen sensitive disease, yet patients with ILC face uniquely poor long-term outcomes with increased recurrence risk, suggesting endocrine response and ER function are unique in ILC. We previously found specifically in ILC cells that ER is co-regulated by the DNA repair protein Mediator of DNA Damage Checkpoint 1 (MDC1). This novel MDC1 activity, however, was associated with dysfunction in the canonical DNA repair activity of MDC1, but absent typical features of DNA repair deficiency. To understand reciprocal activities of MDC1, we profiled the MDC1 interactome and found MDC1-associated proteins in ILC cells mirror a "BRCA-like" state lacking key homologous recombination (HR) proteins, consistent with HR dysfunction but distinct from classic "BRCAness". HR dysfunction in ILC cells was mirrored in single-cell transcriptome and DNA repair activity analyses, along with DNA repair signaling and functional data, showing dysfunctional HR induction and resolution. In parallel, ILC tumor data are consistent with a distinct form of HR dysfunction via impaired HR resolution, lacking BRCA-like genomic scarring but with elevated signatures of PARP inhibitor sensitivity. We tested whether this HR dysfunction could indeed be exploited using PARP inhibition and found that talazoparib treatment produced a durable growth suppression in vitro and in multiple ILC xenografts in vivo. ILC-specific ER:MDC1 activity creates a new context for ER and MDC1 function in ILC, at the cost of a DNA repair dysfunction that is therapeutically targetable.
- Nanopore sequencing of intact aminoacylated tRNAsLaura K White, Aleksandar Radakovic, Marcin P Sajek, and 5 more authorsbioRxiv, Nov 2024
Transfer RNAs (tRNA) are decorated during biogenesis with a variety of modifications that modulate their stability, aminoacylation, and decoding potential during translation. The complex landscape of tRNA modification presents significant analysis challenges and to date no single approach enables the simultaneous measurement of important but disparate chemical properties of individual, mature tRNA molecules. We developed a new, integrated approach to analyze the sequence, modification, and aminoacylation state of tRNA molecules in a high throughput nanopore sequencing experiment, leveraging a chemical ligation that embeds the charged amino acid in an adapted tRNA molecule. During nanopore sequencing, the embedded amino acid generates unique distortions in ionic current and translocation speed, enabling application of machine learning approaches to classify charging status and amino acid identity. Specific applications of the method indicate it will be broadly useful for examining relationships and dependencies between tRNA sequence, modification, and aminoacylation.
-
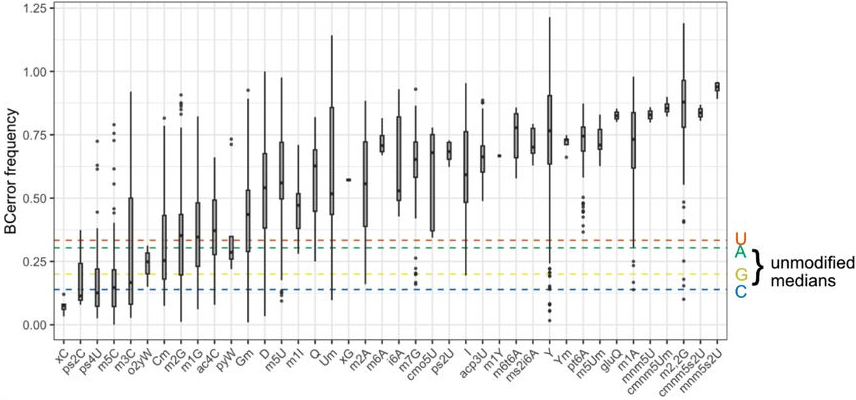 Comparative analysis of 43 distinct RNA modifications by nanopore tRNA sequencingLaura K White, Kezia Dobson, Samantha Pozo, and 10 more authorsJul 2024
Comparative analysis of 43 distinct RNA modifications by nanopore tRNA sequencingLaura K White, Kezia Dobson, Samantha Pozo, and 10 more authorsJul 2024Transfer RNAs are the fundamental adapter molecules of protein synthesis and the most abundant and heterogeneous class of noncoding RNA molecules in cells. The study of tRNA repertoires remains challenging, complicated by the presence of dozens of post transcriptional modifications. Nanopore sequencing is an emerging technology with promise for both tRNA sequencing and the detection of RNA modifications; however, such studies have been limited by the throughput and accuracy of direct RNA sequencing methods. Moreover, detection of the complete set of tRNA modifications by nanopore sequencing remains challenging. Here we show that recent updates to nanopore direct RNA sequencing chemistry (RNA004) combined with our own optimizations to tRNA sequencing protocols and analysis workflows enable high throughput coverage of tRNA molecules and characterization of nanopore signals produced by 43 distinct RNA modifications. We share best practices and protocols for nanopore sequencing of tRNA and further report successful detection of low abundance mitochondrial and viral tRNAs, providing proof of concept for use of nanopore sequencing to study tRNA populations in the context of infection and organelle biology. This work provides a roadmap to guide future efforts towards de novo detection of RNA modifications across multiple organisms using nanopore sequencing.Competing Interest StatementL.K.W. has received travel and accommodation expenses from Oxford Nanopore Technologies to present at scientific meetings, and has been a participant in ONT Early Access programs for SQK-RNA004. The remaining authors declare no conflicts of interest.
-
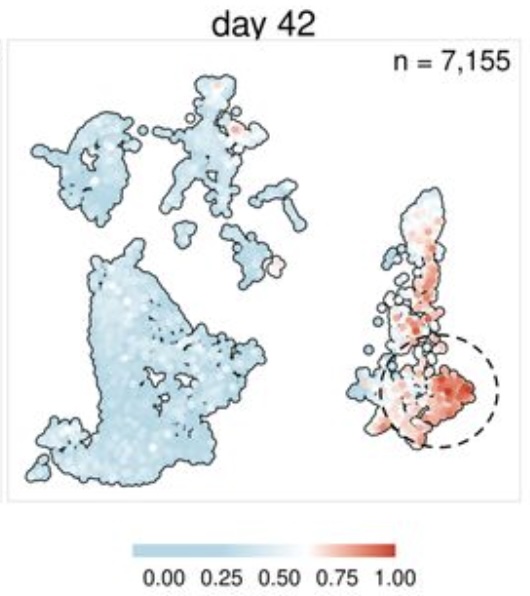 A specific and portable gene expression program underlies antigen archiving by lymphatic endothelial cellsRyan M Sheridan, Thu A Doan, Cormac Lucas, and 5 more authorsApr 2024
A specific and portable gene expression program underlies antigen archiving by lymphatic endothelial cellsRyan M Sheridan, Thu A Doan, Cormac Lucas, and 5 more authorsApr 2024Antigens from protein subunit vaccination traffic from the tissue to the draining lymph node, either passively via the lymph or carried by dendritic cells at the local injection site. Lymph node (LN) lymphatic endothelial cells (LEC) actively acquire and archive foreign antigens, and archived antigen can be released during subsequent inflammatory stimulus to improve immune responses. Here, we answer questions about how LECs achieve durable antigen archiving and whether there are transcriptional signatures associated with LECs containing high levels of antigen. We used single cell sequencing in dissociated LN tissue to quantify antigen levels in LEC and dendritic cell populations at multiple timepoints after immunization, and used machine learning to define a unique transcriptional program within archiving LECs that can predict LEC archiving capacity in independent data sets. Finally, we validated this modeling, showing we could predict antigen archiving from a transcriptional dataset of CHIKV infected mice and demonstrated in vivo the accuracy of our prediction. Collectively, our findings establish a unique transcriptional program in LECs that promotes antigen archiving that can be translated to other systems. ### Competing Interest Statement The authors have declared no competing interest.
- Immunization-induced antigen archiving enhances local memory CD8+ T cell responses following an unrelated viral infectionThu A Doan, Tadg S Forward, Johnathon B Schafer, and 8 more authorsNPJ Vaccines, Mar 2024
Antigens from viruses or immunizations can persist or are archived in lymph node stromal cells such as lymphatic endothelial cells (LEC) and fibroblastic reticular cells (FRC). Here, we find that, during the time frame of antigen archiving, LEC apoptosis caused by a second, but unrelated, innate immune stimulus such as vaccina viral infection or CpG DNA administration resulted in cross-presentation of archived antigens and boosted memory CD8 + T cells specific to the archived antigen. In contrast to “bystander” activation associated with unrelated infections, the memory CD8 + T cells specific to the archived antigen from the immunization were significantly higher than memory CD8 + T cells of a different antigen specificity. Finally, the boosted memory CD8 + T cells resulted in increased protection against Listeria monocytogenes expressing the antigen from the immunization, but only for the duration that the antigen was archived. These findings outline an important mechanism by which lymph node stromal cell archived antigens, in addition to bystander activation, can augment memory CD8 + T cell responses during repeated inflammatory insults.
- Peptidyl-tRNA hydrolysis rate influences the efficiency of nonsense-mediated mRNA decayDivya Kolakada, Rui Fu, Nikita Biziaev, and 9 more authorsbioRxiv, Jan 2024
Nonsense-mediated mRNA decay (NMD) is a quality control mechanism that prevents the accumulation of harmful truncated proteins by degrading transcripts with premature termination codons (PTCs). NMD efficiency varies across many contexts, but the factors that influence this variability remain poorly understood. Here, we find an enrichment of glycine (Gly) codons preceding a PTC in common nonsense variants in contrast with a depletion of Gly codons preceding a normal termination codon (NTC). Gly-PTC contexts have higher NMD activity compared to an alanine-PTC context, and this effect is stronger on NMD substrates with long 3’UTRs. We used a massively parallel reporter assay to test all possible combinations of -2 and -1 codons, the PTC, and the +4 nucleotide to assess comprehensively how PTC sequence context affects NMD efficiency. A random forest classifier revealed that peptidyl-tRNA hydrolysis rate during translation termination was the most important feature in discriminating high and low NMD activity. We show with in vitro biochemical assays that Gly-TC contexts have the slowest termination rate compared to other codons. Furthermore, Gly-PTC enrichment is most pronounced in genes that tolerate loss-of-function variants, suggesting that enhanced NMD of Gly-PTC context has shaped the evolution of PTCs. Based on these findings, we propose that NMD efficiency is modulated by the “window of opportunity” offered by peptidyl tRNA hydrolysis rate and thus, translation termination kinetics.
- Chikungunya virus infection disrupts lymph node lymphatic endothelial cell composition and function via MARCOCormac J Lucas, Ryan M Sheridan, Glennys V Reynoso, and 7 more authorsJCI Insight, Jan 2024
Infection with chikungunya virus (CHIKV) causes disruption of draining lymph node (dLN) organization, including paracortical relocalization of B cells, loss of the B cell-T cell border, and lymphocyte depletion that is associated with infiltration of the LN with inflammatory myeloid cells. Here, we found that, during the first 24 hours of infection, CHIKV RNA accumulated in MARCO-expressing lymphatic endothelial cells (LECs) in both the floor and medullary LN sinuses. The accumulation of viral RNA in the LN was associated with a switch to an antiviral and inflammatory gene expression program across LN stromal cells, and this inflammatory response - including recruitment of myeloid cells to the LN - was accelerated by CHIKV-MARCO interactions. As CHIKV infection progressed, both floor and medullary LECs diminished in number, suggesting further functional impairment of the LN by infection. Consistent with this idea, antigen acquisition by LECs, a key function of LN LECs during infection and immunization, was reduced during pathogenic CHIKV infection.
2023
2023
- Vaccine-induced antigen archiving enhances local memory CD8+ T cell responses following an unrelated viral infectionBeth Tamburini, Thu Doan, Tadg Forward, and 5 more authorsRes Sq, Sep 2023
Viral and vaccine antigens persist or are archived in lymph node stromal cells (LNSC) such as lymphatic endothelial cells (LEC) and fibroblastic reticular cells (FRC). Here, we find that, during the time frame of antigen archiving, LEC apoptosis caused by a second, but unrelated, innate immune stimulus such as vaccina viral infection or CpG DNA administration boosted memory CD8+ T cells specific to the archived antigen. In contrast to “bystander” activation associated with unrelated infections, the memory CD8+ T cells specific to the vaccine archived antigen were significantly higher than memory CD8+ T cells of a different antigen specificity. Finally, the boosted memory CD8+ T cells resulted in increased protection against Listeria monocytogenes expressing the vaccine antigen, but only for the duration that the vaccine antigen was archived. These findings outline a novel mechanism by which LNSC archived antigens, in addition to bystander activation, can augment memory CD8+ T cell responses during repeated inflammatory insults.
- Vaccine adjuvant-elicited CD8+ T cell immunity is co-dependent on T-bet and FOXO1Daria L Ivanova, Scott B Thompson, Jared Klarquist, and 6 more authorsCell Rep., Jul 2023
T-bet and FOXO1 are transcription factors canonically associated with effector and memory T cell fates, respectively. During an infectious response, these factors direct the development of CD8+ T cell fates, where T-bet deficiency leads to ablation of only short-lived effector cells, while FOXO1 deficiency results in selective loss of memory. In contrast, following adjuvanted subunit vaccination in mice, both effector- and memory-fated T cells are compromised in the absence of either T-bet or FOXO1. Thus, unlike responses to challenge with Listeria monocytogenes, productive CD8+ T cell responses to adjuvanted vaccination require coordinated regulation of FOXO1 and T-bet transcriptional programs. Single-cell RNA sequencing analysis confirms simultaneous T-bet, FOXO1, and TCF1 transcriptional activity in vaccine-elicited, but not infection-elicited, T cells undergoing clonal expansion. Collectively, our data show that subunit vaccine adjuvants elicit T cell responses dependent on transcription factors associated with effector and memory cell fates.
-
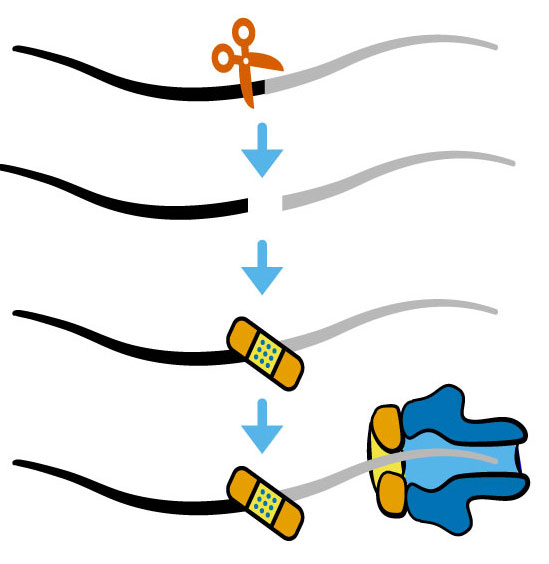 Nanopore sequencing of internal 2’-PO4 modifications installed by RNA repairLaura K White, Saylor M Strugar, Andrea MacFadden, and 1 more authorRNA, Jun 2023
Nanopore sequencing of internal 2’-PO4 modifications installed by RNA repairLaura K White, Saylor M Strugar, Andrea MacFadden, and 1 more authorRNA, Jun 2023Ligation by plant and fungal RNA ligases yields an internal 2’-phosphate group on each RNA ligation product. In budding yeast, this covalent mark occurs at the splice junction of two targets of ligation: intron-containing tRNAs and the messenger RNA HAC1 The repertoire of RNA molecules repaired by RNA ligation has not been explored due to a lack of unbiased approaches for identifying RNA ligation products. Here, we define several unique signals produced by 2’-phosphorylated RNAs during nanopore sequencing. A 2’-phosphate at the splice junction of HAC1 mRNA inhibits 5’ →3’ degradation, enabling detection of decay intermediates in yeast RNA repair mutants by nanopore sequencing. During direct RNA sequencing, intact 2’-phosphorylated RNAs on HAC1 and tRNAs produce diagnostic changes in nanopore current properties and base calling features, including stalls produced as the modified RNA translocates through the nanopore motor protein. These approaches enable directed studies to identify novel RNA repair events in other contexts.
2022
2022
-
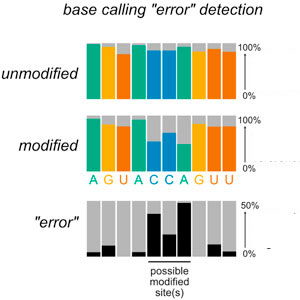 Modification mapping by nanopore sequencingLaura K White and Jay R HesselberthFront. Genet., Oct 2022
Modification mapping by nanopore sequencingLaura K White and Jay R HesselberthFront. Genet., Oct 2022Next generation sequencing (NGS) has provided biologists with an unprecedented view into biological processes and their regulation over the past 2 decades, fueling a wave of development of high throughput methods based on short read DNA and RNA sequencing. For nucleic acid modifications, NGS has been coupled with immunoprecipitation, chemical treatment, enzymatic treatment, and/or the use of reverse transcriptase enzymes with fortuitous activities to enrich for and to identify covalent modifications of RNA and DNA. However, the majority of nucleic acid modifications lack commercial monoclonal antibodies, and mapping techniques that rely on chemical or enzymatic treatments to manipulate modification signatures add additional technical complexities to library preparation. Moreover, such approaches tend to be specific to a single class of RNA or DNA modification, and generate only indirect readouts of modification status. Third generation sequencing technologies such as the commercially available “long read” platforms from Pacific Biosciences and Oxford Nanopore Technologies are an attractive alternative for high throughput detection of nucleic acid modifications. While the former can indirectly sense modified nucleotides through changes in the kinetics of reverse transcription reactions, nanopore sequencing can in principle directly detect any nucleic acid modification that produces a signal distortion as the nucleic acid passes through a nanopore sensor embedded within a charged membrane. To date, more than a dozen endogenous DNA and RNA modifications have been interrogated by nanopore sequencing, as well as a number of synthetic nucleic acid modifications used in metabolic labeling, structure probing, and other emerging applications. This review is intended to introduce the reader to nanopore sequencing and key principles underlying its use in direct detection of nucleic acid modifications in unamplified DNA or RNA samples, and outline current approaches for detecting and quantifying nucleic acid modifications by nanopore sequencing. As this technology matures, we anticipate advances in both sequencing chemistry and analysis methods will lead to rapid improvements in the identification and quantification of these epigenetic marks.
-
 scraps: an end-to-end pipeline for measuring alternative polyadenylation at high resolution using single-cell RNA-seqRui Fu, Kent A Riemondy, Ryan M Sheridan, and 3 more authorsAug 2022
scraps: an end-to-end pipeline for measuring alternative polyadenylation at high resolution using single-cell RNA-seqRui Fu, Kent A Riemondy, Ryan M Sheridan, and 3 more authorsAug 2022Alternative cleavage and polyadenylation (APA) contributes to the diversity of mRNA 3’ ends, affecting post-transcriptional regulation by including or excluding cis -regulatory elements in mRNAs, altering their stability and translational efficiency. While APA analysis has been applied broadly in mixed populations of cells, the heterogeneity of APA among single cells has only recently begun to be explored. We developed an approach we termed scraps (Single Cell RNA PolyA Site Discovery), implemented as a user-friendly, scalable, and reproducible end-to-end workflow, to identify polyadenylation sites at near-nucleotide resolution in single cells using 10X Genomics and other TVN-primed single-cell RNA-seq (scRNA-seq) libraries. Our approach, which performs best with long (>100bp) read 1 sequencing and paired alignment to the genome, is both unbiased relative to existing methods that utilize only read 2 and recovers more sites at higher resolution, despite the reduction in read quality observed on most modern DNA sequencers following homopolymer stretches. For libraries sequenced without long read 1, we implement a fallback approach using read 2-only alignments that performs similarly to our optimal approach, but recovers far fewer polyadenylation sites per experiment. scraps also enables assessment of internal priming capture events, which we demonstrate occur commonly but at higher frequency during apoptotic 3’ RNA decay. We also provide an R package, scrapR, that integrates the results of the scaps pipeline with the popular Seruat single-cell analysis package. Refinement and expanded application of these approaches will further clarify the role of APA in single cells, as well as the effects of internal priming on expression measurements in scRNA-seq libraries. ### Competing Interest Statement The authors have declared no competing interest.
2021
2021
- Haploinsufficiency, Dominant Negative, and Gain-of-Function Mechanisms in Epilepsy: Matching Therapeutic Approach to the PathophysiologyGemma L Carvill, Tyler Matheny, Jay Hesselberth, and 1 more authorNeurotherapeutics, Jul 2021
This review summarizes the pathogenic mechanisms that underpin the monogenic epilepsies and discusses the potential of novel precision therapeutics to treat these disorders. Pathogenic mechanisms of epilepsy include recessive (null alleles), haploinsufficiency, imprinting, gain-of-function, and dominant negative effects. Understanding which pathogenic mechanism(s) that underlie each genetic epilepsy is pivotal to design precision therapies that are most likely to be beneficial for the patient. Novel therapeutics discussed include gene therapy, gene editing, antisense oligonucleotides, and protein replacement. Discussions are illustrated and reinforced with examples from the literature.
- MARCO+ lymphatic endothelial cells sequester arthritogenic alphaviruses to limit viremia and viral disseminationKathryn S Carpentier, Ryan M Sheridan, Cormac J Lucas, and 10 more authorsEMBO J., Oct 2021
Viremia in the vertebrate host is a major determinant of arboviral reservoir competency, transmission efficiency, and disease severity. However, immune mechanisms that control arboviral viremia are poorly defined. Here, we identify critical roles for the scavenger receptor MARCO in controlling viremia during arthritogenic alphavirus infections in mice. Following subcutaneous inoculation, arthritogenic alphavirus particles drain via the lymph and are rapidly captured by MARCO+ lymphatic endothelial cells (LECs) in the draining lymph node (dLN), limiting viral spread to the bloodstream. Upon reaching the bloodstream, alphavirus particles are cleared from the circulation by MARCO-expressing Kupffer cells in the liver, limiting viremia and further viral dissemination. MARCO-mediated accumulation of alphavirus particles in the draining lymph node and liver is an important host defense mechanism as viremia and viral tissue burdens are elevated in MARCO-/- mice and disease is more severe. In contrast to prior studies implicating a key role for lymph node macrophages in limiting viral dissemination, these findings exemplify a previously unrecognized arbovirus-scavenging role for lymphatic endothelial cells and improve our mechanistic understanding of viremia control during arthritogenic alphavirus infection.
- Cell-level metadata are indispensable for documenting single-cell sequencing datasetsSidhant Puntambekar, Jay R Hesselberth, Kent A Riemondy, and 1 more authorPLoS Biol., May 2021
Single-cell RNA sequencing (scRNA-seq) provides an unprecedented view of cellular diversity of biological systems. However, across the thousands of publications and datasets generated using this technology, we estimate that only a minority (<25%) of studies provide cell-level metadata information containing identified cell types and related findings of the published dataset. Metadata omission hinders reproduction, exploration, validation, and knowledge transfer and is a common problem across journals, data repositories, and publication dates. We encourage investigators, reviewers, journals, and data repositories to improve their standards and ensure proper documentation of these valuable datasets.
- Neoplastic and immune single cell transcriptomics define subgroup-specific intra-tumoral heterogeneity of childhood medulloblastomaKent A Riemondy, Sujatha Venkataraman, Nicholas Willard, and 18 more authorsNeuro. Oncol., Jun 2021
BACKGROUND: Medulloblastoma (MB) is a heterogeneous disease in which neoplastic cells and associated immune cells contribute to disease progression. We aimed to determine the influence of neoplastic and immune cell diversity on MB biology in patient samples and animal models. METHODS: To better characterize cellular heterogeneity in MB we used single-cell RNA sequencing, immunohistochemistry and deconvolution of transcriptomic data to profile neoplastic and immune populations in patient samples and animal models across childhood MB subgroups. RESULTS: Neoplastic cells cluster primarily according to individual sample of origin which is influenced by chromosomal copy number variance. Harmony alignment reveals novel MB subgroup/subtype-associated subpopulations that recapitulate neurodevelopmental processes, including photoreceptor and glutamatergic neuron-like cells in molecular subgroups GP3 and GP4, and a specific nodule-associated neuronally-differentiated subpopulation in subgroup molecular SHH. We definitively chart the spectrum of MB immune cell infiltrates, which include subpopulations that recapitulate developmentally-related neuron-pruning and antigen presenting myeloid cells. MB cellular diversity matching human samples is mirrored in subgroup-specific mouse models of MB. CONCLUSIONS: These findings provide a clearer understanding of the diverse neoplastic and immune cell subpopulations that constitute the MB microenvironment.
-
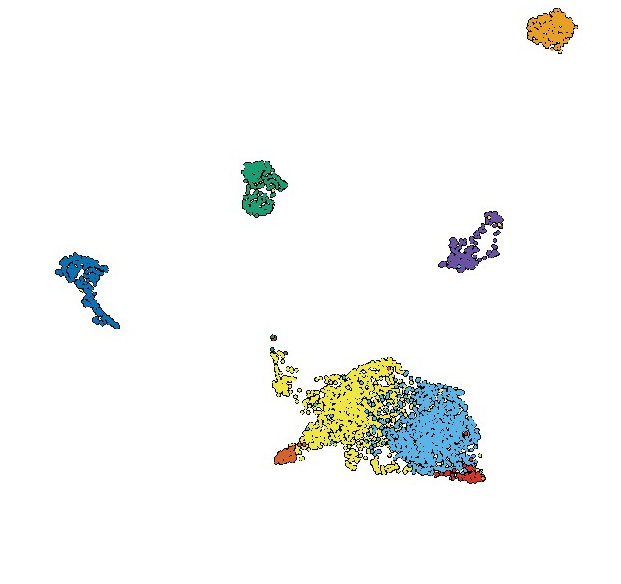 Molecular tracking devices quantify antigen distribution and archiving in the murine lymph nodeShannon M Walsh, Ryan M Sheridan, Erin D Lucas, and 7 more authorsElife, Apr 2021
Molecular tracking devices quantify antigen distribution and archiving in the murine lymph nodeShannon M Walsh, Ryan M Sheridan, Erin D Lucas, and 7 more authorsElife, Apr 2021The detection of foreign antigens in vivo has relied on fluorescent conjugation or indirect read-outs such as antigen presentation. In our studies, we found that these widely used techniques had several technical limitations that have precluded a complete picture of antigen trafficking or retention across lymph node cell types. To address these limitations, we developed a ’molecular tracking device’ to follow the distribution, acquisition, and retention of antigen in the lymph node. Utilizing an antigen conjugated to a nuclease-resistant DNA tag, acting as a combined antigen-adjuvant conjugate, and single-cell mRNA sequencing, we quantified antigen abundance in the lymph node. Variable antigen levels enabled the identification of caveolar endocytosis as a mechanism of antigen acquisition or retention in lymphatic endothelial cells. Thus, these molecular tracking devices enable new approaches to study dynamic tissue dissemination of antigen-adjuvant conjugates and identify new mechanisms of antigen acquisition and retention at cellular resolution in vivo.
2020
2020
- Enhancing Hematopoiesis from Murine Embryonic Stem Cells through MLL1-Induced Activation of a Rac/Rho/Integrin Signaling AxisWeiwei Yang, G Devon Trahan, Elizabeth D Howell, and 7 more authorsStem Cell Reports, Feb 2020
The Mixed Lineage Leukemia (MLL1, KMT2A) gene is critical for development and maintenance of hematopoietic stem cells (HSCs), however, whether this protein is limiting for HSC development is unknown due to lack of physiologic model systems. Here, we develop an MLL1-inducible embryonic stem cell (ESC) system and show that induction of wild-type MLL1 during ESC differentiation selectively increases hematopoietic potential from a transitional c-Kit+/Cd41+ population in the embryoid body and also at sites of hematopoiesis in embryos. Single-cell sequencing analysis illustrates inherent heterogeneity of the c-Kit+/Cd41+ population and demonstrates that MLL1 induction shifts its composition toward multilineage hematopoietic identities. Surprisingly, this does not occur through increasing Hox or other canonical MLL1 targets but through an enhanced Rac/Rho/integrin signaling state, which increases responsiveness to Vla4 ligands and enhances hematopoietic commitment. Together, our data implicate a Rac/Rho/integrin signaling axis in the endothelial to hematopoietic transition and demonstrate that MLL1 actives this axis.
- Dynamic RNA Regulation in the Brain Underlies Physiological Plasticity in a Hibernating MammalRui Fu, Austin E Gillen, Katharine R Grabek, and 5 more authorsFront. Physiol., Feb 2020
Hibernation is a physiological and behavioral phenotype that minimizes energy expenditure. Hibernators cycle between profound depression and rapid hyperactivation of multiple physiological processes, challenging our concept of mammalian homeostasis. How the hibernator orchestrates and survives these extremes while maintaining cell to organismal viability is unknown. Here, we enhance the genome integrity and annotation of a model hibernator, the 13-lined ground squirrel. Our new assembly brings this genome to near chromosome-level contiguity and adds thousands of previously unannotated genes. These new genomic resources were used to identify 6,505 hibernation-related, differentially-expressed and processed transcripts using RNA-seq data from three brain regions in animals whose physiological status was precisely defined using body temperature telemetry. A software tool, squirrelBox, was developed to foster further data analyses and visualization. SquirrelBox includes a comprehensive toolset for rapid visualization of gene level and cluster group dynamics, sequence scanning of k-mer and domains, and interactive exploration of gene lists. Using these new tools and data, we deconvolute seasonal from temperature-dependent effects on the brain transcriptome during hibernation for the first time, highlighting the importance of carefully timed samples for studies of differential gene expression in hibernation. The identified genes include a regulatory network of RNA binding proteins that are dynamic in hibernation along with the composition of the RNA pool. In addition to passive effects of temperature, we provide evidence for regulated transcription and RNA turnover during hibernation. Significant alternative splicing, largely temperature dependent, also occurs during hibernation. These findings form a crucial first step and provide a roadmap for future work toward defining novel mechanisms of tissue protection and metabolic depression that may 1 day be applied toward improving human health.
-
 clustifyr: an R package for automated single-cell RNA sequencing cluster classificationRui Fu, Austin E Gillen, Ryan M Sheridan, and 5 more authorsF1000Res., Apr 2020
clustifyr: an R package for automated single-cell RNA sequencing cluster classificationRui Fu, Austin E Gillen, Ryan M Sheridan, and 5 more authorsF1000Res., Apr 2020Assignment of cell types from single-cell RNA sequencing (scRNA-seq) data remains a time-consuming and error-prone process. Current packages for identity assignment use limited types of reference data and often have rigid data structure requirements. We developed the clustifyr R package to leverage several external data types, including gene expression profiles to assign likely cell types using data from scRNA-seq, bulk RNA-seq, microarray expression data, or signature gene lists. We benchmark various parameters of a correlation-based approach and implement gene list enrichment methods. clustifyr is a lightweight and effective cell-type assignment tool developed for compatibility with various scRNA-seq analysis workflows. clustifyr is publicly available at https://github.com/rnabioco/clustifyr.
-
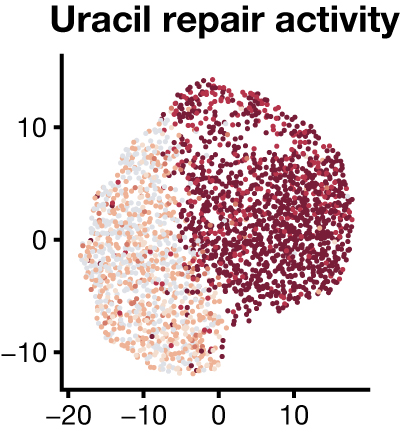 Simultaneous measurement of biochemical phenotypes and gene expression in single cellsAmanda L Richer, Kent A Riemondy, Lakotah Hardie, and 1 more authorNucleic Acids Res., Jun 2020
Simultaneous measurement of biochemical phenotypes and gene expression in single cellsAmanda L Richer, Kent A Riemondy, Lakotah Hardie, and 1 more authorNucleic Acids Res., Jun 2020Methods to measure heterogeneity among cells are rapidly transforming our understanding of biology but are currently limited to molecular abundance measurements. We developed an approach to simultaneously measure biochemical activities and mRNA abundance in single cells to understand the heterogeneity of DNA repair activities across thousands of human lymphocytes, identifying known and novel cell-type-specific DNA repair phenotypes.
- Physiologic RNA targets and refined sequence specificity of coronavirus EndoURachel Ancar, Yize Li, Eveline Kindler, and 6 more authorsRNA, Dec 2020
Coronavirus EndoU inhibits dsRNA-activated antiviral responses; however, the physiologic RNA substrates of EndoU are unknown. In this study, we used mouse hepatitis virus (MHV)-infected bone marrow-derived macrophage (BMM) and cyclic phosphate cDNA sequencing to identify the RNA targets of EndoU. EndoU targeted viral RNA, cleaving the 3’ side of pyrimidines with a strong preference for U A and C A sequences (endoY A). EndoU-dependent cleavage was detected in every region of MHV RNA, from the 5’ NTR to the 3’ NTR, including transcriptional regulatory sequences (TRS). Cleavage at two CA dinucleotides immediately adjacent to the MHV poly(A) tail suggests a mechanism to suppress negative-strand RNA synthesis and the accumulation of viral dsRNA. MHV with EndoU (EndoUmut) or 2’-5’ phosphodiesterase (PDEmut) mutations provoked the activation of RNase L in BMM, with corresponding cleavage of RNAs by RNase L. The physiologic targets of EndoU are viral RNA templates required for negative-strand RNA synthesis and dsRNA accumulation. Coronavirus EndoU cleaves U A and C A sequences (endoY A) within viral (+) strand RNA to evade dsRNA-activated host responses.
- Single-Cell RNA Sequencing of Childhood Ependymoma Reveals Neoplastic Cell Subpopulations That Impact Molecular Classification and EtiologyAustin E Gillen, Kent A Riemondy, Vladimir Amani, and 14 more authorsCell Rep., Aug 2020
Ependymoma (EPN) is a brain tumor commonly presenting in childhood that remains fatal in most children. Intra-tumoral cellular heterogeneity in bulk-tumor samples significantly confounds our understanding of EPN biology, impeding development of effective therapy. We, therefore, use single-cell RNA sequencing, histology, and deconvolution to catalog cellular heterogeneity of the major childhood EPN subgroups. Analysis of PFA subgroup EPN reveals evidence of an undifferentiated progenitor subpopulation that either differentiates into subpopulations with ependymal cell characteristics or transitions into a mesenchymal subpopulation. Histological analysis reveals that progenitor and mesenchymal subpopulations co-localize in peri-necrotic zones. In conflict with current classification paradigms, relative PFA subpopulation proportions are shown to determine bulk-tumor-assigned subgroups. We provide an interactive online resource that facilitates exploration of the EPN single-cell dataset. This atlas of EPN cellular heterogeneity increases understanding of EPN biology.
-
 Custom-designed, degradation-resistant messenger RNAs in yeastAna L Franklin, Andrea Macfadden, Jeffrey S Kieft, and 2 more authorsJun 2020
Custom-designed, degradation-resistant messenger RNAs in yeastAna L Franklin, Andrea Macfadden, Jeffrey S Kieft, and 2 more authorsJun 2020Structured RNA elements that protect RNA transcripts from 5’\rightarrow3’ degradation are becoming useful research tools. Here we show that exonuclease-resistant RNA structures (xrRNAs) from Flaviviruses can be used to protect heterologous messenger RNAs (mRNAs) from 5’\rightarrow3’ degradation in budding yeast. Installation of xrRNAs ahead of a downstream internal ribosome entry site (IRES) leads to the accumulation of partially-degraded mRNAs that are substrates for cap-independent translation of a LacZ reporter, yielding a 30-fold increase in measured β-galactosidase activity. Additionally, by monitoring the translation of dual-luciferase reporters we show that xrRNA sequences do not interfere with the progression of an elongating ribosome. Combined these data indicate that xrRNA elements can be used in creative ways to stabilize RNAs with potentially useful applications. ### Competing Interest Statement AM, JSK, JRH and EGC are listed as inventors of PCT/US2016/066723 that describes Protecting RNAs From Degradation Using Engineered Viral RNAs. This patent is owned by the Reagents of the University of Colorado.
- Single cell profiling identifies novel B cell populations that drive chronic rejection after lung transplantationN Smirnova, K Riemondy, S Collins, and 2 more authorsJ. Heart Lung Transplant., Apr 2020
2019
2019
- Addendum: Ribose-seq: global mapping of ribonucleotides embedded in genomic DNAKyung Duk Koh, Sathya Balachander, Jay R Hesselberth, and 1 more authorNat. Methods, Aug 2019
- METHODS TO MEASURE FUNCTIONAL HETEROGENEITY AMONG SINGLE CELLSA L Richer and J R HesselberthAug 2019
Methods of measuring multiple enzyme activities in parallel in a sequencing-based assay to characterize enzyme activities in individual mammalian cells. In preferred implementations, the methods involve forming microfluidic droplets containing oligonucleotide functionalized microbeads and single mammalian cells, lysing the cells, and allowing enzyme activity on enzyme substrates present in the oligonucleotides, isolating the individual microbeads, and determining the enzymatic activity to quantitate and evaluate the enzymatic activity or capacity of the cells.
- Single-cell RNAseq of childhood ependymoma reveals distinct neoplastic cell subpopulations that impact etiology, molecular classification and outcomeAustin Gillen, Kent Riemondy, Vladimir Amani, and 14 more authorsSSRN Electron. J., Aug 2019
- Chronic liver disease in humans causes expansion and differentiation of liver lymphatic endothelial cellsBeth A Jiron Tamburini, Jeffrey M Finlon, Austin E Gillen, and 7 more authorsFront. Immunol., May 2019
Liver lymphatic vessels support liver function by draining interstitial fluid, cholesterol, fat, and immune cells for surveillance in the liver draining lymph node. Chronic liver disease is associated with increased inflammation and immune cell infiltrate. However, it is currently unknown if or how lymphatic vessels respond to increased inflammation and immune cell infiltrate in the liver during chronic disease. Here we demonstrate that lymphatic vessel abundance increases in patients with chronic liver disease and is associated with areas of fibrosis and immune cell infiltration. Using single-cell mRNA sequencing and multi-spectral immunofluorescence analysis we identified liver lymphatic endothelial cells and found that chronic liver disease results in lymphatic endothelial cells (LECs) that are in active cell cycle with increased expression of CCL21. Additionally, we found that LECs from patients with NASH adopt a transcriptional program associated with increased IL13 signaling. Moreover, we found that oxidized low density lipoprotein, associated with NASH pathogenesis, induced the transcription and protein production of IL13 in LECs both in vitro and in a mouse model. Finally, we show that oxidized low density lipoprotein reduced the transcription of PROX1 and decreased lymphatic stability. Together these data indicate that LECs are active participants in the liver, expanding in an attempt to maintain tissue homeostasis. However, when inflammatory signals, such as oxidized low density lipoprotein are increased, as in NASH, lymphatic function declines and liver homeostasis is impeded.
- Single cell RNA sequencing identifies TGFβ as a key regenerative cue following LPS-induced lung injuryKent A Riemondy, Nicole L Jansing, Peng Jiang, and 8 more authorsJCI Insight, Mar 2019
Many lung diseases result from a failure of efficient regeneration of damaged alveolar epithelial cells (AECs) after lung injury. During regeneration, AEC2s proliferate to replace lost cells, after which proliferation halts and some AEC2s transdifferentiate into AEC1s to restore normal alveolar structure and function. Although the mechanisms underlying AEC2 proliferation have been studied, the mechanisms responsible for halting proliferation and inducing transdifferentiation are poorly understood. To identify candidate signaling pathways responsible for halting proliferation and inducing transdifferentiation, we performed single cell RNA sequencing on AEC2s during regeneration in a murine model of lung injury induced by intratracheal LPS. Unsupervised clustering revealed distinct subpopulations of regenerating AEC2s: proliferating, cell cycle arrest, and transdifferentiating. Gene expression analysis of these transitional subpopulations revealed that TGFβsignaling was highly upregulated in the cell cycle arrest subpopulation and relatively downregulated in transdifferentiating cells. In cultured AEC2s, TGFβwas necessary for cell cycle arrest but impeded transdifferentiation. We conclude that during regeneration after LPS-induced lung injury, TGFβis a critical signal halting AEC2 proliferation but must be inactivated to allow transdifferentiation. This study provides insight into the molecular mechanisms regulating alveolar regeneration and the pathogenesis of diseases resulting from a failure of regeneration.
- Recovery and analysis of transcriptome subsets from pooled single-cell RNA-seq librariesKent A Riemondy, Monica Ransom, Christopher Alderman, and 8 more authorsNucleic Acids Res., Feb 2019
Single-cell RNA sequencing (scRNA-seq) methods generate sparse gene expression profiles for thousands of single cells in a single experiment. The information in these profiles is sufficient to classify cell types by distinct expression patterns but the high complexity of scRNA-seq libraries often prevents full characterization of transcriptomes from individual cells. To extract more focused gene expression information from scRNA-seq libraries, we developed a strategy to physically recover the DNA molecules comprising transcriptome subsets, enabling deeper interrogation of the isolated molecules by another round of DNA sequencing. We applied the method in cell-centric and gene-centric modes to isolate cDNA fragments from scRNA-seq libraries. First, we resampled the transcriptomes of rare, single megakaryocytes from a complex mixture of lymphocytes and analyzed them in a second round of DNA sequencing, yielding up to 20-fold greater sequencing depth per cell and increasing the number of genes detected per cell from a median of 1313 to 2002. We similarly isolated mRNAs from targeted T cells to improve the reconstruction of their VDJ-rearranged immune receptor mRNAs. Second, we isolated CD3D mRNA fragments expressed across cells in a scRNA-seq library prepared from a clonal T cell line, increasing the number of cells with detected CD3D expression from 59.7% to 100%. Transcriptome resampling is a general approach to recover targeted gene expression information from single-cell RNA sequencing libraries that enhances the utility of these costly experiments, and may be applicable to the targeted recovery of molecules from other single-cell assays.
-
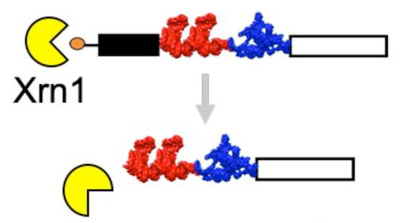
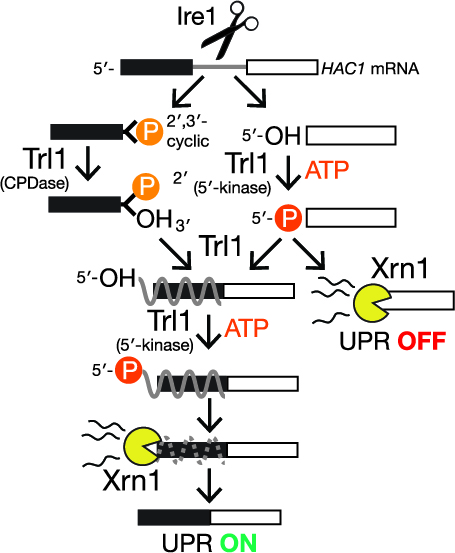 Multiple decay events target HAC1 mRNA during splicing to regulate the unfolded protein responsePatrick D Cherry, Sally E Peach, and Jay R HesselberthElife, Mar 2019
Multiple decay events target HAC1 mRNA during splicing to regulate the unfolded protein responsePatrick D Cherry, Sally E Peach, and Jay R HesselberthElife, Mar 2019In the unfolded protein response (UPR), stress in the endoplasmic reticulum (ER) activates a large transcriptional program to increase ER folding capacity. During the budding yeast UPR, Ire1 excises an intron from the HAC1 mRNA and the exon products of cleavage are ligated, and the translated protein induces hundreds of stress-response genes. Using cells with mutations in RNA repair and decay enzymes, we show that phosphorylation of two different HAC1 splicing intermediates is required for their degradation by the 5’\rightarrow3’ exonuclease Xrn1 to enact opposing effects on the UPR. We also found that ligated but 2’-phosphorylated HAC1 mRNA is cleaved, yielding a decay intermediate with both 5’- and 2’-phosphates at its 5’-end that inhibit 5’\rightarrow3’ decay and suggesting that Ire1 degrades incompletely processed HAC1. These decay events expand the scope of RNA-based regulation in the budding yeast UPR and have implications for the control of the metazoan UPR.
2018
2018
- O Cdc7 kinase where art thou?Robert A Sclafani and Jay R HesselberthCurr. Genet., Jun 2018
Although Cdc7 protein kinase is important for regulating DNA replication in all eukaryotes and is a target for cancer therapy, it has never been localized in cells. Recently, a novel molecular genomic method used by our laboratory to localize Cdc7 to regions of chromosomes. Originally, mutations in the CDC7 gene were found in the classic cdc mutant collection of Hartwell et al. (Genetics 74:267-286, 1973). The CDC7 gene was found to encode a protein kinase called DDK that has been studied for many years, establishing its precise role in the initiation of DNA replication at origins. Recently, clinical studies are underway with DDK inhibitors against DDK in cancer patients. However, the conundrum is that Cdc7 has never been detected at origins of replication even though many studies have suggested it should be there. We used “Calling Card” system in which DNA binding proteins are localized to the genome via retrotransposon insertion and deep-sequencing methods. We have shown that Cdc7 localizes at many regions of the genome and was enriched at functional origins of replication. These results are consistent with DDK’s role in many additional genomic processes including mutagenesis, chromatid cohesion, and meiotic recombination. Thus, the main conclusion from our studies is that Cdc7 kinase is found at many locations in the genome, but is enriched at functional origins of replication. Furthermore, we propose that application of the Calling Card system to other eukaryotes should be useful in identification of functional origins in other eukaryotic cells.
- High-Resolution Mapping of Modified DNA Nucleobases Using Excision Repair EnzymesMonica Ransom, D Suzi Bryan, and Jay R HesselberthMethods Mol. Biol., Jun 2018
Modification of DNA nucleobases has a profound effect on genome function. We developed a method that maps the positions of the modified DNA nucleobases throughout genomic DNA. This method couples in vitro nucleobase excision with massively parallel DNA sequencing to determine the location of modified DNA nucleobases with single base precision. This protocol was used to map uracil incorporation and UV photodimers in DNA, and a modification of the protocol has been used to map sparse modification events in cells. The Excision-seq protocol is broadly applicable to a variety of base modifications for which an excision enzyme is available.
- TCR signal strength controls thymic differentiation of iNKT cell subsetsKathryn D Tuttle, S Harsha Krovi, Jingjing Zhang, and 13 more authorsNat. Commun., Jul 2018
During development in the thymus, invariant natural killer T (iNKT) cells commit to one of three major functionally different subsets, iNKT1, iNKT2, and iNKT17. Here, we show that T cell antigen receptor (TCR) signal strength governs the development of iNKT cell subsets, with strong signaling promoting iNKT2 and iNKT17 development. Altering TCR diversity or signaling diminishes iNKT2 and iNKT17 cell subset development in a cell-intrinsic manner. Decreased TCR signaling affects the persistence of Egr2 expression and the upregulation of PLZF. By genome-wide comparison of chromatin accessibility, we identify a subset of iNKT2-specific regulatory elements containing NFAT and Egr binding motifs that is less accessible in iNKT2 cells that develop from reduced TCR signaling. These data suggest that variable TCR signaling modulates regulatory element activity at NFAT and Egr binding sites exerting a determinative influence on the dynamics of gene enhancer accessibility and the developmental fate of iNKT cells.
- Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemiaDaniel A Pollyea, Brett M Stevens, Courtney L Jones, and 14 more authorsNat. Med., Dec 2018
Acute myeloid leukemia (AML) is the most common acute leukemia in adults. Leukemia stem cells (LSCs) drive the initiation and perpetuation of AML, are quantifiably associated with worse clinical outcomes, and often persist after conventional chemotherapy resulting in relapse1-5. In this report, we show that treatment of older patients with AML with the B cell lymphoma 2 (BCL-2) inhibitor venetoclax in combination with azacitidine results in deep and durable remissions and is superior to conventional treatments. We hypothesized that these promising clinical results were due to targeting LSCs. Analysis of LSCs from patients undergoing treatment with venetoclax + azacitidine showed disruption of the tricarboxylic acid (TCA) cycle manifested by decreased α-ketoglutarate and increased succinate levels, suggesting inhibition of electron transport chain complex II. In vitro modeling confirmed inhibition of complex II via reduced glutathionylation of succinate dehydrogenase. These metabolic perturbations suppress oxidative phosphorylation (OXPHOS), which efficiently and selectively targets LSCs. Our findings show for the first time that a therapeutic intervention can eradicate LSCs in patients with AML by disrupting the metabolic machinery driving energy metabolism, resulting in promising clinical activity in a patient population with historically poor outcomes.
- Dynamic temperature-sensitive A-to-I RNA editing in the brain of a heterothermic mammal during hibernationKent A Riemondy, Austin E Gillen, Emily A White, and 3 more authorsRNA, Nov 2018
RNA editing diversifies genomically encoded information to expand the complexity of the transcriptome. In ectothermic organisms, including Drosophila and Cephalopoda, where body temperature mirrors ambient temperature, decreases in environmental temperature lead to increases in A-to-I RNA editing and cause amino acid recoding events that are thought to be adaptive responses to temperature fluctuations. In contrast, endothermic mammals, including humans and mice, typically maintain a constant body temperature despite environmental changes. Here, A-to-I editing primarily targets repeat elements, rarely results in the recoding of amino acids, and plays a critical role in innate immune tolerance. Hibernating ground squirrels provide a unique opportunity to examine RNA editing in a heterothermic mammal whose body temperature varies over 30C and can be maintained at 5C for many days during torpor. We profiled the transcriptome in three brain regions at six physiological states to quantify RNA editing and determine whether cold-induced RNA editing modifies the transcriptome as a potential mechanism for neuroprotection at low temperature during hibernation. We identified 5165 A-to-I editing sites in 1205 genes with dynamically increased editing after prolonged cold exposure. The majority (99.6%) of the cold-increased editing sites are outside of previously annotated coding regions, 82.7% lie in SINE-derived repeats, and 12 sites are predicted to recode amino acids. Additionally, A-to-I editing frequencies increase with increasing cold-exposure, demonstrating that ADAR remains active during torpor. Our findings suggest that dynamic A-to-I editing at low body temperature may provide a neuroprotective mechanism to limit aberrant dsRNA accumulation during torpor in the mammalian hibernator.
- TCR signal strength controls thymic differentiation of iNKT cell subsetsKathryn D Tuttle, S Harsha Krovi, Jingjing Zhang, and 13 more authorsNat. Commun., Jul 2018
During development in the thymus, invariant natural killer T (iNKT) cells commit to one of three major functionally different subsets, iNKT1, iNKT2, and iNKT17. Here, we show that T cell antigen receptor (TCR) signal strength governs the development of iNKT cell subsets, with strong signaling promoting iNKT2 and iNKT17 development. Altering TCR diversity or signaling diminishes iNKT2 and iNKT17 cell subset development in a cell-intrinsic manner. Decreased TCR signaling affects the persistence of Egr2 expression and the upregulation of PLZF. By genome-wide comparison of chromatin accessibility, we identify a subset of iNKT2-specific regulatory elements containing NFAT and Egr binding motifs that is less accessible in iNKT2 cells that develop from reduced TCR signaling. These data suggest that variable TCR signaling modulates regulatory element activity at NFAT and Egr binding sites exerting a determinative influence on the dynamics of gene enhancer accessibility and the developmental fate of iNKT cells.
-
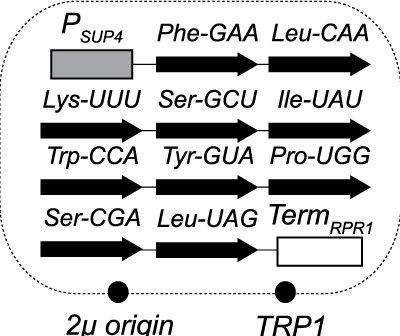 Genetic bypass of essential RNA repair enzymes in budding yeastPatrick D Cherry, Laura K White, Kerri York, and 1 more authorRNA, Mar 2018
Genetic bypass of essential RNA repair enzymes in budding yeastPatrick D Cherry, Laura K White, Kerri York, and 1 more authorRNA, Mar 2018RNA repair enzymes catalyze rejoining of an RNA molecule after cleavage of phosphodiester linkages. RNA repair in budding yeast is catalyzed by two separate enzymes that process tRNA exons during their splicing andHAC1mRNA exons during activation of the unfolded protein response (UPR). The RNA ligase Trl1 joins 2’,3’-cyclic phosphate and 5’-hydroxyl RNA fragments, creating a phosphodiester linkage with a 2’-phosphate at the junction. The 2’-phosphate is removed by the 2’-phosphotransferase Tpt1. We bypassed the essential functions ofTRL1andTPT1in budding yeast by expressing “prespliced,” intronless versions of the 10 normally intron-containing tRNAs, indicating this repair pathway does not have additional essential functions. Consistent with previous studies, expression of intronless tRNAs failed to rescue the growth of cells with deletions in components of the SEN complex, implying an additional essential role for the splicing endonuclease. Thetrl1∆andtpt1∆mutants accumulate tRNA andHAC1splicing intermediates indicative of RNA repair defects and are hypersensitive to drugs that inhibit translation. Failure to induce the unfolded protein response intrl1∆cells grown with tunicamycin is lethal owing to their inability to ligateHAC1after its cleavage by Ire1. In contrast,tpt1∆mutants grow in the presence of tunicamycin despite reduced accumulation of splicedHAC1mRNA. We optimized a PCR-based method to detect RNA 2’-phosphate modifications and show they are present on ligatedHAC1mRNA. These RNA repair mutants enable new studies of the role of RNA repair in cellular physiology.
2017
2017
- Localization of Cdc7 Protein Kinase During DNA Replication in Saccharomyces cerevisiaeDaniel Rossbach, D Suzi Bryan, Jay R Hesselberth, and 1 more authorG3, Nov 2017
DDK, a conserved serine-threonine protein kinase composed of a regulatory subunit, Dbf4, and a catalytic subunit, Cdc7, is essential for DNA replication initiation during S phase of the cell cycle through MCM2-7 helicase phosphorylation. The biological significance of DDK is well characterized, but the full mechanism of how DDK associates with substrates remains unclear. Cdc7 is bound to chromatin in the Saccharomyces cerevisiae genome throughout the cell cycle, but there is little empirical evidence as to specific Cdc7 binding locations. Using biochemical and genetic techniques, this study investigated the specific localization of Cdc7 on chromatin. The Calling Cards method, using Ty5 retrotransposons as a marker for DNA-protein binding, suggests Cdc7 kinase is preferentially bound to genomic DNA known to replicate early in S phase, including centromeres and origins of replication. We also discovered Cdc7 binding throughout the genome, which may be necessary to initiate other cellular processes, including meiotic recombination and translesion synthesis. A kinase dead Cdc7 point mutation increases the Ty5 retrotransposon integration efficiency and a 55-amino acid C-terminal truncation of Cdc7, unable to bind Dbf4, reduces Cdc7 binding suggesting a requirement for Dbf4 to stabilize Cdc7 on chromatin during S phase. Chromatin immunoprecipitation demonstrates that Cdc7 binding near specific origins changes during S phase. Our results suggest a model where Cdc7 is loosely bound to chromatin during G1 At the G1/S transition, Cdc7 binding to chromatin is increased and stabilized, preferentially at sites that may become origins, in order to carry out a variety of cellular processes.
- Alternative Polyadenylation of PRELID1 Regulates Mitochondrial ROS Signaling and Cancer OutcomesAustin E Gillen, Heather M Brechbuhl, Tomomi M Yamamoto, and 4 more authorsMol. Cancer Res., Dec 2017
Disruption of posttranscriptional gene regulation is a critical step in oncogenesis that can be difficult to observe using traditional molecular techniques. To overcome this limitation, a modified polyadenylation site sequencing (PAS-seq) protocol was used to generate a genome-wide map of alternative polyadenylation (APA) events in human primary breast tumor specimens and matched normal tissue. This approach identified an APA event in the PRELID1 mRNA that enhances its steady-state level and translational efficiency, and is a strong breast cancer subtype-dependent predictor of patient clinical outcomes. Furthermore, it has been demonstrated that PRELID1 regulates stress response and mitochondrial reactive oxygen species (ROS) production in a cell type-specific manner. Modulation of PRELID1 expression, including its posttranscriptional control, appears to be a common stress response across different cancer types. These data reveal that PRELID1 mRNA processing is an important regulator of cell type-specific responses to stress used by multiple cancers and is associated with patient outcomes.Implications: This study suggests that the regulation of PRELID1 expression, by APA and other mechanisms, plays a role in mitochondrial ROS signaling and represents a novel prognostic factor and therapeutic target in cancer. Mol Cancer Res; 15(12); 1741-51. \copyright2017 AACR.
-
 valr: Reproducible genome interval analysis in RKent A Riemondy, Ryan M Sheridan, Austin Gillen, and 3 more authorsF1000Res., Jun 2017
valr: Reproducible genome interval analysis in RKent A Riemondy, Ryan M Sheridan, Austin Gillen, and 3 more authorsF1000Res., Jun 2017New tools for reproducible exploratory data analysis of large datasets are important to address the rising size and complexity of genomic data. We developed the valr R package to enable flexible and efficient genomic interval analysis. valr leverages new tools available in the “tidyverse”, including dplyr. Benchmarks of valr show it performs similar to BEDtools and can be used for interactive analyses and incorporated into existing analysis pipelines.
2016
2016
- Improvements to the HITS-CLIP protocol eliminate widespread mispriming artifactsAustin E Gillen, Tomomi M Yamamoto, Enos Kline, and 2 more authorsBMC Genomics, May 2016
BACKGROUND: High-throughput sequencing of RNA isolated by crosslinking immunoprecipitation (HITS-CLIP) allows for high resolution, genome-wide mapping of RNA-binding proteins. This methodology is frequently used to validate predicted targets of microRNA binding, as well as direct targets of other RNA-binding proteins. Hence, the accuracy and sensitivity of binding site identification is critical. RESULTS: We found that substantial mispriming during reverse transcription results in the overrepresentation of sequences complementary to the primer used for reverse transcription. Up to 45 % of peaks in publicly available HITS-CLIP libraries are attributable to this mispriming artifact, and the majority of libraries have detectable levels of mispriming. We also found that standard techniques for validating microRNA-target interactions fail to differentiate between artifactual peaks and physiologically relevant peaks. CONCLUSIONS: Here, we present a modification to the HITS-CLIP protocol that effectively eliminates this artifact and improves the sensitivity and complexity of resulting libraries.
- Diverse fates of uracilated HIV-1 DNA during infection of myeloid lineage cellsErik C Hansen, Monica Ransom, Jay R Hesselberth, and 9 more authorsElife, Sep 2016
We report that a major subpopulation of monocyte-derived macrophages (MDMs) contains high levels of dUTP, which is incorporated into HIV-1 DNA during reverse transcription (U/A pairs), resulting in pre-integration restriction and post-integration mutagenesis. After entering the nucleus, uracilated viral DNA products are degraded by the uracil base excision repair (UBER) machinery with less than 1% of the uracilated DNA successfully integrating. Although uracilated proviral DNA showed few mutations, the viral genomic RNA was highly mutated, suggesting that errors occur during transcription. Viral DNA isolated from blood monocytes and alveolar macrophages (but not T cells) of drug-suppressed HIV-infected individuals also contained abundant uracils. The presence of viral uracils in short-lived monocytes suggests their recent infection through contact with virus producing cells in a tissue reservoir. These findings reveal new elements of a viral defense mechanism involving host UBER that may be relevant to the establishment and persistence of HIV-1 infection.
2015
2015
- Coordination between Drosophila Arc1 and a specific population of brain neurons regulates organismal fatJeremy Mosher, Wei Zhang, Rachel Z Blumhagen, and 5 more authorsDev. Biol., Sep 2015
The brain plays a critical yet incompletely understood role in regulating organismal fat. We performed a neuronal silencing screen in Drosophila larvae to identify brain regions required to maintain proper levels of organismal fat. When used to modulate synaptic activity in specific brain regions, the enhancer-trap driver line E347 elevated fat upon neuronal silencing, and decreased fat upon neuronal activation. Unbiased sequencing revealed that Arc1 mRNA levels increase upon E347 activation. We had previously identified Arc1 mutations in a high-fat screen. Here we reveal metabolic changes in Arc1 mutants consistent with a high-fat phenotype and an overall shift toward energy storage. We find that Arc1-expressing cells neighbor E347 neurons, and manipulating E347 synaptic activity alters Arc1 expression patterns. Elevating Arc1 expression in these cells decreased fat, a phenocopy of E347 activation. Finally, loss of Arc1 prevented the lean phenotype caused by E347 activation, suggesting that Arc1 activity is required for E347 control of body fat. Importantly, neither E347 nor Arc1 manipulation altered energy-related behaviors. Our results support a model wherein E347 neurons induce Arc1 in specific neighboring cells to prevent excess fat accumulation.
- Enhanced stability and polyadenylation of select mRNAs support rapid thermogenesis in the brown fat of a hibernatorKatharine R Grabek, Cecilia Diniz Behn, Gregory S Barsh, and 2 more authorsElife, Jan 2015
During hibernation, animals cycle between torpor and arousal. These cycles involve dramatic but poorly understood mechanisms of dynamic physiological regulation at the level of gene expression. Each cycle, Brown Adipose Tissue (BAT) drives periodic arousal from torpor by generating essential heat. We applied digital transcriptome analysis to precisely timed samples to identify molecular pathways that underlie the intense activity cycles of hibernator BAT. A cohort of transcripts increased during torpor, paradoxical because transcription effectively ceases at these low temperatures. We show that this increase occurs not by elevated transcription but rather by enhanced stabilization associated with maintenance and/or extension of long poly(A) tails. Mathematical modeling further supports a temperature-sensitive mechanism to protect a subset of transcripts from ongoing bulk degradation instead of increased transcription. This subset was enriched in a C-rich motif and genes required for BAT activation, suggesting a model and mechanism to prioritize translation of key proteins for thermogenesis.
- RNase L targets distinct sites in influenza A virus RNAsDaphne A Cooper, Shuvojit Banerjee, Arindam Chakrabarti, and 4 more authorsJ. Virol., Mar 2015
UNLABELLED: Influenza A virus (IAV) infections are influenced by type 1 interferon-mediated antiviral defenses and by viral countermeasures to these defenses. When IAV NS1 protein is disabled, RNase L restricts virus replication; however, the RNAs targeted for cleavage by RNase L under these conditions have not been defined. In this study, we used deep-sequencing methods to identify RNase L cleavage sites within host and viral RNAs from IAV PR8\DeltaNS1-infected A549 cells. Short hairpin RNA knockdown of RNase L allowed us to distinguish between RNase L-dependent and RNase L-independent cleavage sites. RNase L-dependent cleavage sites were evident at discrete locations in IAV RNA segments (both positive and negative strands). Cleavage in PB2, PB1, and PA genomic RNAs suggests that viral RNPs are susceptible to cleavage by RNase L. Prominent amounts of cleavage mapped to specific regions within IAV RNAs, including some areas of increased synonymous-site conservation. Among cellular RNAs, RNase L-dependent cleavage was most frequent at precise locations in rRNAs. Our data show that RNase L targets specific sites in both host and viral RNAs to restrict influenza virus replication when NS1 protein is disabled. IMPORTANCE: RNase L is a critical component of interferon-regulated and double-stranded-RNA-activated antiviral host responses. We sought to determine how RNase L exerts its antiviral activity during influenza virus infection. We enhanced the antiviral activity of RNase L by disabling a viral protein, NS1, that inhibits the activation of RNase L. Then, using deep-sequencing methods, we identified the host and viral RNAs targeted by RNase L. We found that RNase L cleaved viral RNAs and rRNAs at very precise locations. The direct cleavage of IAV RNAs by RNase L highlights an intimate battle between viral RNAs and an antiviral endonuclease.
- Global analysis of RNA cleavage by 5’-hydroxyl RNA sequencingSally E Peach, Kerri York, and Jay R HesselberthNucleic Acids Res., Sep 2015
RNA cleavage by some endoribonucleases and self-cleaving ribozymes produces RNA fragments with 5’-hydroxyl (5’-OH) and 2’,3’-cyclic phosphate termini. To identify 5’-OH RNA fragments produced by these cleavage events, we exploited the unique ligation mechanism of Escherichia coli RtcB RNA ligase to attach an oligonucleotide linker to RNAs with 5’-OH termini, followed by steps for library construction and analysis by massively parallel DNA sequencing. We applied the method to RNA from budding yeast and captured known 5’-OH fragments produced by tRNA Splicing Endonuclease (SEN) during processing of intron-containing pre-tRNAs and by Ire1 cleavage of HAC1 mRNA following induction of the unfolded protein response (UPR). We identified numerous novel 5’-OH fragments derived from mRNAs: some 5’-OH mRNA fragments were derived from single, localized cleavages, while others were likely produced by multiple, distributed cleavages. Many 5’-OH fragments derived from mRNAs were produced upstream of codons for highly electrostatic peptides, suggesting that the fragments may be generated by co-translational mRNA decay. Several 5’-OH RNA fragments accumulated during the induction of the UPR, some of which share a common sequence motif that may direct cleavage of these mRNAs. This method enables specific capture of 5’-OH termini and complements existing methods for identifying RNAs with 2’,3’-cyclic phosphate termini.
- Ribose-seq: global mapping of ribonucleotides embedded in genomic DNAKyung Duk Koh, Sathya Balachander, Jay R Hesselberth, and 1 more authorNat. Methods, Mar 2015
Abundant ribonucleotide incorporation in DNA during replication and repair has profound consequences for genome stability, but the global distribution of ribonucleotide incorporation is unknown. We developed ribose-seq, a method for capturing unique products generated by alkaline cleavage of DNA at embedded ribonucleotides. High-throughput sequencing of these fragments in DNA from the yeast Saccharomyces cerevisiae revealed widespread ribonucleotide distribution, with a strong preference for cytidine and guanosine, and identified hotspots of ribonucleotide incorporation in nuclear and mitochondrial DNA. Ribonucleotides were primarily incorporated on the newly synthesized leading strand of nuclear DNA and were present upstream of (G+C)-rich tracts in the mitochondrial genome. Ribose-seq is a powerful tool for the systematic profiling of ribonucleotide incorporation in genomic DNA.
- Global analysis of RNA cleavage by 5’-hydroxyl RNA sequencingSally E Peach, Kerri York, and Jay R HesselberthNucleic Acids Res., Sep 2015
RNA cleavage by some endoribonucleases and self-cleaving ribozymes produces RNA fragments with 5’-hydroxyl (5’-OH) and 2’,3’-cyclic phosphate termini. To identify 5’-OH RNA fragments produced by these cleavage events, we exploited the unique ligation mechanism of Escherichia coli RtcB RNA ligase to attach an oligonucleotide linker to RNAs with 5’-OH termini, followed by steps for library construction and analysis by massively parallel DNA sequencing. We applied the method to RNA from budding yeast and captured known 5’-OH fragments produced by tRNA Splicing Endonuclease (SEN) during processing of intron-containing pre-tRNAs and by Ire1 cleavage of HAC1 mRNA following induction of the unfolded protein response (UPR). We identified numerous novel 5’-OH fragments derived from mRNAs: some 5’-OH mRNA fragments were derived from single, localized cleavages, while others were likely produced by multiple, distributed cleavages. Many 5’-OH fragments derived from mRNAs were produced upstream of codons for highly electrostatic peptides, suggesting that the fragments may be generated by co-translational mRNA decay. Several 5’-OH RNA fragments accumulated during the induction of the UPR, some of which share a common sequence motif that may direct cleavage of these mRNAs. This method enables specific capture of 5’-OH termini and complements existing methods for identifying RNAs with 2’,3’-cyclic phosphate termini.
2014
2014
- A homolog of lariat-debranching enzyme modulates turnover of branched RNAStephen M Garrey, Adam Katolik, Mantas Prekeris, and 7 more authorsSep 2014
- A homolog of lariat-debranching enzyme modulates turnover of branched RNAStephen M Garrey, Adam Katolik, Mantas Prekeris, and 7 more authorsRNA, Aug 2014
Turnover of the branched RNA intermediates and products of pre-mRNA splicing is mediated by the lariat-debranching enzyme Dbr1. We characterized a homolog of Dbr1 from Saccharomyces cerevisiae, Drn1/Ygr093w, that has a pseudo-metallophosphodiesterase domain with primary sequence homology to Dbr1 but lacks essential active site residues found in Dbr1. Whereas loss of Dbr1 results in lariat-introns failing broadly to turnover, loss of Drn1 causes low levels of lariat-intron accumulation. Conserved residues in the Drn1 C-terminal CwfJ domains, which are not present in Dbr1, are required for efficient intron turnover. Drn1 interacts with Dbr1, components of the Nineteen Complex, U2 snRNA, branched intermediates, and products of splicing. Drn1 enhances debranching catalyzed by Dbr1 in vitro, but does so without significantly improving the affinity of Dbr1 for branched RNA. Splicing carried out in in vitro extracts in the absence of Drn1 results in an accumulation of branched splicing intermediates and products released from the spliceosome, likely due to less active debranching, as well as the promiscuous release of cleaved 5’-exon. Drn1 enhances Dbr1-mediated turnover of lariat-intermediates and lariat-intron products, indicating that branched RNA turnover is regulated at multiple steps during splicing.
- The RtcB RNA ligase is an essential component of the metazoan unfolded protein responseSara Guckian Kosmaczewski, Tyson James Edwards, Sung Min Han, and 6 more authorsEMBO Rep., Aug 2014
- High resolution mapping of modified DNA nucleobases using excision repair enzymesD Suzi Bryan, Monica Ransom, Biniam Adane, and 2 more authorsGenome Res., Sep 2014
The incorporation and creation of modified nucleobases in DNA have profound effects on genome function. We describe methods for mapping positions and local content of modified DNA nucleobases in genomic DNA. We combined in vitro nucleobase excision with massively parallel DNA sequencing (Excision-seq) to determine the locations of modified nucleobases in genomic DNA. We applied the Excision-seq method to map uracil in E. coli and budding yeast and discovered significant variation in uracil content, wherein uracil is excluded from the earliest and latest replicating regions of the genome, possibly driven by changes in nucleotide pool composition. We also used Excision-seq to identify sites of pyrimidine dimer formation induced by UV light exposure, where the method could distinguish between sites of cyclobutane and 6-4 photoproduct formation. These UV mapping data enabled analysis of local sequence bias around pyrimidine dimers and suggested a preference for an adenosine downstream from 6-4 photoproducts. The Excision-seq method is broadly applicable for high precision, genome-wide mapping of modified nucleobases with cognate repair enzymes.
- Ribonuclease L and metal-ion-independent endoribonuclease cleavage sites in host and viral RNAsDaphne A Cooper, Babal K Jha, Robert H Silverman, and 2 more authorsNucleic Acids Res., Apr 2014
Ribonuclease L (RNase L) is a metal-ion-independent endoribonuclease associated with antiviral and antibacterial defense, cancer and lifespan. Despite the biological significance of RNase L, the RNAs cleaved by this enzyme are poorly defined. In this study, we used deep sequencing methods to reveal the frequency and location of RNase L cleavage sites within host and viral RNAs. To make cDNA libraries, we exploited the 2’, 3’-cyclic phosphate at the end of RNA fragments produced by RNase L and other metal-ion-independent endoribonucleases. We optimized and validated 2’, 3’-cyclic phosphate cDNA synthesis and Illumina sequencing methods using viral RNAs cleaved with purified RNase L, viral RNAs cleaved with purified RNase A and RNA from uninfected and poliovirus-infected HeLa cells. Using these methods, we identified (i) discrete regions of hepatitis C virus and poliovirus RNA genomes that were profoundly susceptible to RNase L and other single-strand specific endoribonucleases, (ii) RNase L-dependent and RNase L-independent cleavage sites within ribosomal RNAs (rRNAs) and (iii) 2’, 3’-cyclic phosphates at the ends of 5S rRNA and U6 snRNA. Monitoring the frequency and location of metal-ion-independent endoribonuclease cleavage sites within host and viral RNAs reveals, in part, how these enzymes contribute to health and disease.
- The RtcB RNA ligase is an essential component of the metazoan unfolded protein responseSara Guckian Kosmaczewski, Tyson James Edwards, Sung Min Han, and 6 more authorsEMBO Rep., Dec 2014
RNA ligation can regulate RNA function by altering RNA sequence, structure and coding potential. For example, the function of XBP1 in mediating the unfolded protein response requires RNA ligation, as does the maturation of some tRNAs. Here, we describe a novel in vivo model in Caenorhabditis elegans for the conserved RNA ligase RtcB and show that RtcB ligates the xbp-1 mRNA during the IRE-1 branch of the unfolded protein response. Without RtcB, protein stress results in the accumulation of unligated xbp-1 mRNA fragments, defects in the unfolded protein response, and decreased lifespan. RtcB also ligates endogenous pre-tRNA halves, and RtcB mutants have defects in growth and lifespan that can be bypassed by expression of pre-spliced tRNAs. In addition, animals that lack RtcB have defects that are independent of tRNA maturation and the unfolded protein response. Thus, RNA ligation by RtcB is required for the function of multiple endogenous target RNAs including both xbp-1 and tRNAs. RtcB is uniquely capable of performing these ligation functions, and RNA ligation by RtcB mediates multiple essential processes in vivo.
2013
2013
- Lives that introns lead after splicingJay R HesselberthWiley Interdiscip. Rev. RNA, Nov 2013
After transcription of a eukaryotic pre-mRNA, its introns are removed by the spliceosome, joining exons for translation. The intron products of splicing have long been considered ’junk’ and destined only for destruction. But because they are large in size and under weak selection constraints, many introns have been evolutionarily repurposed to serve roles after splicing. Some spliced introns are precursors for further processing of other encoded RNAs such as small nucleolar RNAs, microRNAs, and long noncoding RNAs. Other intron products have long half-lives and can be exported to the cytoplasm, suggesting that they have roles in translation. Some viruses encode introns that accumulate after splicing and play important but mysterious roles in viral latency. Turnover of most lariat-introns is initiated by cleavage of their internal 2’-5’ phosphodiester bonds by a unique debranching endonuclease, and the linear products are further degraded by exoribonucleases. However, several introns appear to evade this turnover pathway and the determinants of their stability are largely unknown. Whereas many stable intron products were discovered serendipitously, new experimental and computational tools will enable their direct identification and study. Finally, the origins and mechanisms of mobility of eukaryotic introns are mysterious, and mechanistic studies of the intron life cycle may yield new insights into how they arose and became widespread.
2010
2010
- Capture and sequence analysis of RNAs with terminal 2’,3’-cyclic phosphatesKevin Schutz, Jay R Hesselberth, and Stanley FieldsRNA, Mar 2010
The combination of ligation-based RNA capture methods and high-throughput sequencing has facilitated the characterization of transcriptomes and the identification of novel noncoding RNAs. However, current ligation-based RNA capture methods require RNA substrates with terminal 3’-hydroxyl groups, limiting their utility for identifying RNAs with modified termini like 2’,3’-cyclic phosphates. Cyclic phosphate-terminated RNAs are generated by endonucleolytic cleavages and self-cleaving ribozymes and are found as stable modifications on cellular RNAs such as the U6 spliceosomal RNA. We developed a method that uses the Arabidopsis thaliana tRNA ligase to add an adaptor oligonucleotide to RNAs that terminate in 2’,3’-cyclic phosphates. The adaptor allows specific priming by reverse transcriptase, which is followed by additional steps for PCR amplification and high-throughput DNA sequencing. Applying the method to total human RNA, we found 2836 sequencing reads corresponding to the 3’ terminus of U6 snRNA, validating the method. In addition to a large background of reads that map throughout abundantly transcribed RNAs, we also found 42,324 reads of specific fragments from several tRNA isoacceptor families, suggesting that this method may identify processing events previously undetected by other RNA cloning techniques.
- High-throughput profiling of amino acids in strains of the Saccharomyces cerevisiae deletion collectionS J Cooper, G L Finney, S L Brown, and 4 more authorsMar 2010
- Capture and sequence analysis of RNAs with terminal 2’,3’-cyclic phosphatesKevin Schutz, Jay R Hesselberth, and Stanley FieldsRNA, Mar 2010
The combination of ligation-based RNA capture methods and high-throughput sequencing has facilitated the characterization of transcriptomes and the identification of novel noncoding RNAs. However, current ligation-based RNA capture methods require RNA substrates with terminal 3’-hydroxyl groups, limiting their utility for identifying RNAs with modified termini like 2’,3’-cyclic phosphates. Cyclic phosphate-terminated RNAs are generated by endonucleolytic cleavages and self-cleaving ribozymes and are found as stable modifications on cellular RNAs such as the U6 spliceosomal RNA. We developed a method that uses the Arabidopsis thaliana tRNA ligase to add an adaptor oligonucleotide to RNAs that terminate in 2’,3’-cyclic phosphates. The adaptor allows specific priming by reverse transcriptase, which is followed by additional steps for PCR amplification and high-throughput DNA sequencing. Applying the method to total human RNA, we found 2836 sequencing reads corresponding to the 3’ terminus of U6 snRNA, validating the method. In addition to a large background of reads that map throughout abundantly transcribed RNAs, we also found 42,324 reads of specific fragments from several tRNA isoacceptor families, suggesting that this method may identify processing events previously undetected by other RNA cloning techniques.
2009
2009
- Global mapping of protein-DNA interactions in vivo by digital genomic footprintingJay R Hesselberth, Xiaoyu Chen, Zhihong Zhang, and 9 more authorsNat. Methods, Apr 2009
The orchestrated binding of transcriptional activators and repressors to specific DNA sequences in the context of chromatin defines the regulatory program of eukaryotic genomes. We developed a digital approach to assay regulatory protein occupancy on genomic DNA in vivo by dense mapping of individual DNase I cleavages from intact nuclei using massively parallel DNA sequencing. Analysis of >23 million cleavages across the Saccharomyces cerevisiae genome revealed thousands of protected regulatory protein footprints, enabling de novo derivation of factor binding motifs and the identification of hundreds of new binding sites for major regulators. We observed striking correspondence between single-nucleotide resolution DNase I cleavage patterns and protein-DNA interactions determined by crystallography. The data also yielded a detailed view of larger chromatin features including positioned nucleosomes flanking factor binding regions. Digital genomic footprinting should be a powerful approach to delineate the cis-regulatory framework of any organism with an available genome sequence.
2007
2007
- Genome-wide identification of spliced introns using a tiling microarrayZ Zhang, J R Hesselberth, and S FieldsApr 2007
2006
2006
- Binding specificity of Toll-like receptor cytoplasmic domainsVictoria Brown, Rachel A Brown, Adrian Ozinsky, and 2 more authorsEur. J. Immunol., Apr 2006
- Comparative analysis of Saccharomyces cerevisiae WW domains and their interacting proteinsJ R Hesselberth, J P Miller, A Golob, and 2 more authorsGenome Biol., Apr 2006
Background The WW domain is found in a large number of eukaryotic proteins implicated in a variety of cellular processes. WW domains bind proline-rich protein and peptide ligands, but the protein interaction partners of many WW domain-containing proteins in
2005
2005
- A protein interaction network of the malaria parasite Plasmodium falciparumDouglas J LaCount, Marissa Vignali, Rakesh Chettier, and 9 more authorsNature, Nov 2005
Plasmodium falciparum causes the most severe form of malaria and kills up to 2.7 million people annually. Despite the global importance of P. falciparum, the vast majority of its proteins have not been characterized experimentally. Here we identify P. falciparum protein-protein interactions using a high-throughput version of the yeast two-hybrid assay that circumvents the difficulties in expressing P. falciparum proteins in Saccharomyces cerevisiae. From more than 32,000 yeast two-hybrid screens with P. falciparum protein fragments, we identified 2,846 unique interactions, most of which include at least one previously uncharacterized protein. Informatic analyses of network connectivity, coexpression of the genes encoding interacting fragments, and enrichment of specific protein domains or Gene Ontology annotations were used to identify groups of interacting proteins, including one implicated in chromatin modification, transcription, messenger RNA stability and ubiquitination, and another implicated in the invasion of host cells. These data constitute the first extensive description of the protein interaction network for this important human pathogen.
- A protein interaction network of the malaria parasite Plasmodium falciparumDouglas J LaCount, Marissa Vignali, Rakesh Chettier, and 9 more authorsNature, Nov 2005
Plasmodium falciparum causes the most severe form of malaria and kills up to 2.7 million people annually. Despite the global importance of P. falciparum, the vast majority of its proteins have not been characterized experimentally. Here we identify P. falciparum protein-protein interactions using a high-throughput version of the yeast two-hybrid assay that circumvents the difficulties in expressing P. falciparum proteins in Saccharomyces cerevisiae. From more than 32,000 yeast two-hybrid screens with P. falciparum protein fragments, we identified 2,846 unique interactions, most of which include at least one previously uncharacterized protein. Informatic analyses of network connectivity, coexpression of the genes encoding interacting fragments, and enrichment of specific protein domains or Gene Ontology annotations were used to identify groups of interacting proteins, including one implicated in chromatin modification, transcription, messenger RNA stability and ubiquitination, and another implicated in the invasion of host cells. These data constitute the first extensive description of the protein interaction network for this important human pathogen.
- A protein interaction network of the malaria parasite Plasmodium falciparumDouglas J LaCount, Marissa Vignali, Rakesh Chettier, and 9 more authorsNature, Nov 2005
Plasmodium falciparum causes the most severe form of malaria and kills up to 2.7 million people annually. Despite the global importance of P. falciparum, the vast majority of its proteins have not been characterized experimentally. Here we identify P. falciparum protein-protein interactions using a high-throughput version of the yeast two-hybrid assay that circumvents the difficulties in expressing P. falciparum proteins in Saccharomyces cerevisiae. From more than 32,000 yeast two-hybrid screens with P. falciparum protein fragments, we identified 2,846 unique interactions, most of which include at least one previously uncharacterized protein. Informatic analyses of network connectivity, coexpression of the genes encoding interacting fragments, and enrichment of specific protein domains or Gene Ontology annotations were used to identify groups of interacting proteins, including one implicated in chromatin modification, transcription, messenger RNA stability and ubiquitination, and another implicated in the invasion of host cells. These data constitute the first extensive description of the protein interaction network for this important human pathogen.
2004
2004
- Aptamer databaseJennifer F Lee, Jay R Hesselberth, Lauren Ancel Meyers, and 1 more authorNucleic Acids Res., Jan 2004
The aptamer database is designed to contain comprehensive sequence information on aptamers and unnatural ribozymes that have been generated by in vitro selection methods. Such data are not normally collected in ’natural’ sequence databases, such as GenBank. Besides serving as a storehouse of sequences that may have diagnostic or therapeutic utility, the database serves as a valuable resource for theoretical biologists who describe and explore fitness landscapes. The database is updated monthly and is publicly available at http://aptamer. icmb.utexas.edu/.
2003
2003
- Simultaneous detection of diverse analytes with an aptazyme ligase arrayJay R Hesselberth, Michael P Robertson, Scott M Knudsen, and 1 more authorAnal. Biochem., Jan 2003
Allosteric ribozymes (aptazymes) can transduce the noncovalent recognition of analytes into the catalytic generation of readily observable signals. Aptazymes are easily engineered, can detect diverse classes of biologically relevant molecules, and have high signal-to-noise ratios. These features make aptazymes useful candidates for incorporation into biosensor arrays. Allosteric ribozyme ligases that can recognize a variety of analytes ranging from small organics to proteins have been generated. Upon incorporation into an array format, multiple different aptazyme ligases were able to simultaneously detect their cognate analytes with high specificity. Analyte concentrations could be accurately measured into the nanomolar range. The fact that analytes induced the formation of new covalent bonds in aptazyme ligases (as opposed to noncovalent bonds in antibodies) potentiated stringent washing of the array, leading to improved signal-to-noise ratios and limits of detection.
2002
2002
- Automated selection of aptamers against protein targets translated in vitro: from gene to aptamerJ Colin Cox, Andrew Hayhurst, Jay Hesselberth, and 3 more authorsNucleic Acids Res., Oct 2002
Reagents for proteome research must of necessity be generated by high throughput methods. Aptamers are potentially useful as reagents to identify and quantitate individual proteins, yet are currently produced for the most part by manual selection procedures. We have developed automated selection methods, but must still individually purify protein targets. Therefore, we have attempted to select aptamers against protein targets generated by in vitro transcription and translation of individual genes. In order to specifically immobilize the protein targets for selection, they are also biotinylated in vitro. As a proof of this method, we have selected aptamers against translated human U1A, a component of the nuclear spliceosome. Selected sequences demonstrated exquisite mimicry of natural binding sequences and structures. These results not only reveal a potential path to the high throughput generation of aptamers, but also yield insights into the incredible specificity of the U1A protein for its natural RNA ligands.
2001
2001
- Optimization and optimality of a short ribozyme ligase that joins non-Watson–Crick base pairingsMichael P Robertson, Jay R Hesselberth, and Andrew D EllingtonOct 2001
- Optimization and optimality of a short ribozyme ligase that joins non-Watson-Crick base pairingsM P Robertson, J R Hesselberth, and A D EllingtonRNA, Apr 2001
A small ribozyme ligase (L1) selected from a random sequence population appears to utilize non-Watson-Crick base pairs at its ligation junction. Mutational and selection analyses confirmed the presence of these base pairings. Randomization of the L1 core and selection of active ligases yielded highly active variants whose rates were on the order of 1 min(-1). Base-pairing covariations confirmed the general secondary structure of the ligase, and the most active ligases contained a novel pentuple sequence covariation. The optimized L1 ligases may be optimal within their sequence spaces, and minimal ligases that span less than 60 nt in length have been engineered based on these results.
2000
2000
- In vitro selection of nucleic acids for diagnostic applicationsJ Hesselberth, M P Robertson, S Jhaveri, and 1 more authorJ. Biotechnol., Mar 2000
In vitro selection methods have proven to be extraordinarily adept at generating a wide variety of nucleic acid-binding species (aptamers) and catalysts (ribozymes). To date, selected nucleic acids have primarily been of academic interest. However, just as antibodies have proven utility as ’universal receptors’ that can be crafted against a huge variety of ligands and can be readily adapted to diagnostic assays, aptamers may yet find application in assays. A new class of research reagents, aptazymes, are not mere mimics of antibodies but in fact allow the direct transduction of molecular recognition to catalysis. Aptamers and aptazymes may prove to be uniquely useful for the development of chip arrays for the detection and quantitation of a wide range of molecules in organismal proteomes and metabolomes.
- In vitro selection of RNA molecules that inhibit the activity of ricin A-chainJ R Hesselberth, D Miller, J Robertus, and 1 more authorJ. Biol. Chem., Feb 2000
The cytotoxin ricin disables translation by depurinating a conserved site in eukaryotic rRNA. In vitro selection has been used to generate RNA ligands (aptamers) specific for the catalytic ricin A-chain (RTA). The anti-RTA aptamers bear no resemblance to the normal RTA substrate, the sarcin-ricin loop (SRL), and were not depurinated by RTA. An initial 80-nucleotide RNA ligand was minimized to a 31-nucleotide aptamer that contained all sequences and structures necessary for interacting with RTA. This minimal RNA formed high affinity complexes with RTA (K(d) = 7.3 nM) which could compete directly with the SRL for binding to RTA. The aptamer inhibited RTA depurination of the SRL and could partially protect translation from RTA inhibition. The IC(50) of the aptamer for RTA in an in vitro translation assay is 100 nM, roughly 3 orders of magnitude lower than a small molecule inhibitor of ricin, pteroic acid, and 2 orders of magnitude lower than the best known RNA inhibitor. The novel anti-RTA aptamers may find application as diagnostic reagents for a potential biological warfare agent and hold promise as scaffolds for the development of strong ricin inhibitors.